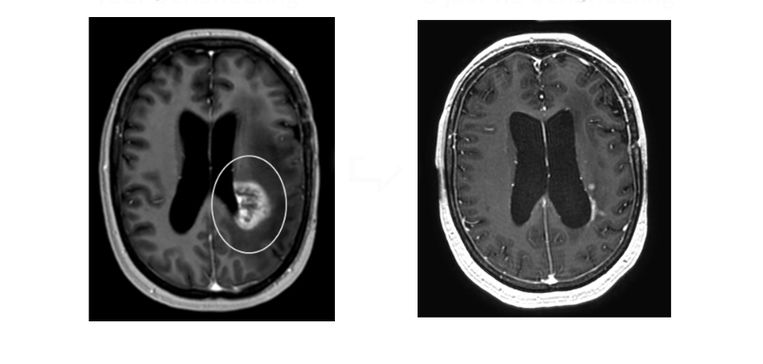

Registration and abstract submission are now open for the ESGCT 30th Annual Congress. This year’s Congress is being held in collaboration with the SFTCG and NVGCT and takes place from 24-27 October in Brussels, Belgium.
Free registration is offered to student members who submit an abstract which is accepted for an oral or poster presentation. Register by 30 June to get Early Bird rates, plus reduced registration fees will apply to all ESGCT members. NVGCT members are entitled to free membership of the ESGCT for 2023, which can be requested via events@esgct.eu with proof of your NVGCT membership. This proof of NVGCT membership can be acquired via info@nvgct.nl.
Abstract submission deadline: 15 June 2023.